

Jak firma manipulowała wynikami badań COVID aby uzyskać zgodę FDA?


Analiza i porównanie dokumentu przeglądowego przedłożonego przez firmę Pfizer do amerykańskiej Agencji ds. Żywności i Leków, na podstawie którego FDA dała zielone światło na rozszerzenie zezwolenia na szczepienie w sytuacjach nagłych również dla dzieci w wieku 12-15 lat (1), w przeciwieństwie do wyników badania u dzieci (2, 3) ujawnia ustalenia, w tym naruszenia wyników ustalonego przez samą firmę Pfizer, a nie mniej poważne, zaprojektowanie protokołu badania w taki sposób, aby firma mogła przedstawić jak najbardziej pozytywne wyniki w warunki bezpieczeństwa szczepionek u dzieci.
Zgodnie z dokumentem przeglądowym przedłożonym przez firmę Pfizer do FDA, czworo z 1131 dzieci w ramieniu badania, które otrzymały szczepionkę Pfizer-BioNTech COVID-19, doznało poważnych zdarzeń niepożądanych („SAE") – tj. zdarzeń, w których co najmniej jedno kryterium: zgon spowodowany, zagraża życiu, wymaga hospitalizacji lub przedłużenia dotychczasowej hospitalizacji, skutkuje trwałą inwalidztwem/niezdolnością do pracy lub wadą wrodzoną/wadą wrodzoną.
Spośród tych czworga dzieci troje miało tak ciężką depresję, że były hospitalizowane wkrótce po szczepieniu (w ciągu pierwszych 7 dni po pierwszej dawce, drugie tylko jeden dzień po drugiej dawce, a w trzecim 15 dni po pierwszej dawce, odpowiednio).

Aby nas uspokoić, firma Pfizer zauważa w swoim dokumencie przeglądowym, że w rzeczywistości wszystkie troje dzieci miały wcześniej diagnozę lęku i depresji. Co więcej, wyjaśniają – wszystkie trzy faktycznie zgłaszały selektywny inhibitor wychwytu zwrotnego serotoniny (SSRI), który rozpoczął się w ciągu 1-2 miesięcy przed szczepieniem.
„Pogorszenie myśli samobójczych przy początkowym leczeniu SSRI u nastolatków", wyjaśniają, „jest uznanym ryzykiem i stanowi rozsądne alternatywne wyjaśnienie zaostrzenia depresji u tych biorców BNT162b2".
Jaki jest problem z tym wyjaśnieniem?
Dwa problemy:
- Twierdzenie, że SSRI, które otrzymały dzieci, jest alternatywnym wyjaśnieniem pogorszenia stanu psychicznego dzieci, jest wątpliwe. Według literatury naukowej zaostrzenie myśli o samobójstwie i stanu psychicznego następuje już na początku leczenia antydepresantami, zwykle w pierwszych dwóch tygodniach, a na pewno nie później niż miesiąc po rozpoczęciu leczenia – czyli wtedy, gdy zaczyna się widzieć poprawę . W rzeczywistości jest odwrotnie: jeśli nie ma poprawy w ciągu czterech tygodni, zwykle zastępuje się lek.
- Co ważniejsze, zgodnie z wynikami badania – uczestnicy z wcześniejszą diagnozą psychiatryczną nigdy nie powinni byli być włączani do badania w pierwszej kolejności (patrz str. 41 w protokole). Okazuje się, że jednym z kryteriów wykluczających z badania jest: „Inny stan chorobowy lub psychiatryczny, w tym niedawne (w ciągu ostatniego roku) lub aktywne myśli/zachowania samobójcze lub nieprawidłowe wyniki laboratoryjne, które mogą zwiększać ryzyko udziału w badaniu lub, w osąd, uczynić uczestnika nieodpowiednim do badania".

Zaprojektowanie badań w sposób, który umożliwi firmie przedstawienie pozytywnych ustaleń dotyczących bezpieczeństwa szczepionki
Wydaje się, że w co najmniej dwóch kryteriach firma manipulująco zaprojektowała protokół w sposób, który umożliwiłby przedstawienie jak najbardziej pozytywnych wyników w zakresie bezpieczeństwa szczepionek u dzieci:
1.Zaprojektowanie protokołu w taki sposób, aby maksymalnie ograniczyć uwzględnianie poważnych zdarzeń niepożądanych w raporcie przedkładanym FDA.
W protokole badań pediatrycznych (patrz tabela na stronie 12) firma Pfizer zobowiązała się, że okres obserwacji w przypadku poważnych zdarzeń niepożądanych (SAE) będzie wynosił „od dawki 1 do 6 miesięcy po podaniu drugiej dawki".
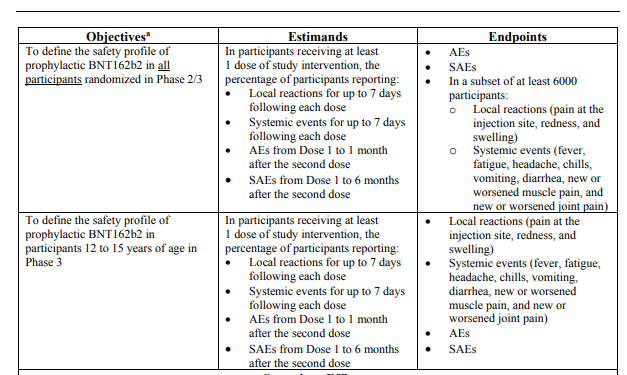
Jednak z dokumentu przeglądowego firmy Pfizer wynika, że firma nie ukończyła nawet tego stosunkowo krótkiego okresu obserwacji i faktycznie była zadowolona z zaledwie 30 dni obserwacji poważnych zdarzeń niepożądanych. Fakt ten wynika z rozdziału dotyczącego daty analizy (strona 30, pod nagłówkiem rozdziału SAE): „Dzieci w wieku 12-15 lat: SAE od Dawki 1 do 30 dni po Dawce 2 w ciągłej obserwacji wzrost odnotowało 0,4% biorców BNT162b2 i 0,1% biorców placebo".
Na stronie 114 protokołu badania – w rozdziale poświęconym terminom, w jakich będą przeprowadzane analizy statystyczne, firma Pfizer ustaliła liczbę punktów czasowych w celu wykonania tych analiz. Podczas gdy maksymalny okres monitorowania ciężkich zdarzeń niepożądanych w ogólnej populacji badania wynosi sześć miesięcy (siódma sekcja), piąta sekcja ustanowiła dodatkowy punkt odcięcia, wynoszący zaledwie 30 dni po drugiej dawce w celu porównania danych między dwie grupy wiekowe – jedna w wieku 12-15 i jedna w wieku 16-25.

Rzeczywiście, obserwacja poważnych zdarzeń niepożądanych trwa przez kolejne pięć miesięcy, ale każde zdarzenie niepożądane, które zostanie wykryte w ciągu tych miesięcy, lub zdarzenie niepożądane, które zaobserwowano w ciągu pierwszego miesiąca, ale zostało zdefiniowane jako niegroźne i uległo pogorszeniu w ciągu kolejnych miesięcy (albo diagnoza się zmieni) – po prostu nie pojawi się w raporcie z przeglądu.
Niepokojącą konsekwencją tej praktyki jest to, że poważne zdarzenia niepożądane mogą nie pojawić się w raporcie, na podstawie którego FDA wydaje zezwolenie w nagłych wypadkach dla dzieci, więc dalsze działania następcze, nawet jeśli zostały opublikowane kilka miesięcy lub lat po tym, jak tymczasowe zezwolenie zostało wydane, nie pomogą dzieciom, które zostaną skrzywdzone lub umrą po zielonym świetle ze strony FDA.
Zaprojektowanie protokołu w taki sposób, aby można było zignorować diagnozy poważnych zdarzeń niepożądanych postawione w szpitalach niezwiązanych z ośrodkiem badawczym.
Zgodnie z warunkami pomiaru wyników w protokole badania, przedstawionym w Clinicaltrials.gov, firma Pfizer ustaliła, że zespół badawczy wybrany przez firmę Pfizer będzie tym, który definiuje zdarzenia niepożądane jako takie: „Wywołane przez personel ośrodka badawczego".
W ten sposób firma w efekcie dała wybranym przez siebie badaczom możliwość samodzielnego określenia, jaka będzie diagnoza, niezależnie od diagnozy postawionej w szpitalu/oddziale, który nie jest zdefiniowany jako ośrodek badawczy.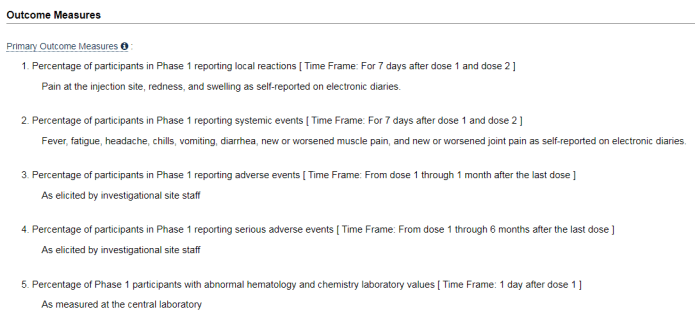
Ponieważ takie ustalenie oznacza, że jeśli u danego uczestnika wystąpiły poważne zdarzenia niepożądane i był leczony np. poza szpitalem lub oddziałem pełniącym funkcję ośrodka badawczego, to w rzeczywistości diagnoza postawiona przez lekarzy prowadzących w szpitalu/ oddział, w którym leczony jest uczestnik, nie ma znaczenia.
W ten sposób firma Pfizer rzeczywiście pozwoliła swojemu zespołowi określić, jaka będzie diagnoza, zamiast pozwolić, aby diagnoza postawiona przez lekarzy prowadzących ich myliła.
Poza ostrą krytyką pod adresem firmy Pfizer, analiza i porównanie rodzą poważne pytania dla samej FDA:
- Jak to możliwe, że FDA w ogóle zatwierdziła protokół, który dopuszcza takie manipulacje?
- Dlaczego FDA zezwoliła firmie na przeprowadzenie analizy danych i złożenie wniosku o wydanie zezwolenia awaryjnego na szczepienie dzieci po tak krótkim czasie obserwacji, wynoszącym zaledwie 30 dni?
- Co sprawiło, że FDA tak chętnie zatwierdziła pozwolenie na nagłe przypadki NOP dla dzieci? Dlaczego ta aprobata jest wydana na podstawie raportu bezpieczeństwa, który nie jest nawet „gotowy" w połowie? W końcu nie ma sytuacji awaryjnej dla dzieci.
- Dlaczego FDA nie zajęła się tymi manipulacjami i naruszeniami protokołu po tym, jak firma przedstawiła swoją recenzję?
Dr Yaffa Shir-Raz jest badaczem komunikacji ryzyka i wykładowcą na Uniwersytecie w Hajfie oraz w Interdyscyplinarnym Centrum Herzliya w Izraelu.
Ten artykuł został opublikowany w American Frontline Doctors.
Bibliografia:
1. Pfizer-BioNTech. (2021). Poprawka do zezwolenia na stosowanie w sytuacjach awaryjnych (EUA) do protokołu przeglądu niezatwierdzonego produktu. https://www.fda.gov/media/148542/download
2. (2021). Faza A 1/2/3 kontrolą placebo randomizowane obserwatora ślepej próby, WYKRYWANIE badanie w celu oceny bezpieczeństwa, tolerancji, immunogenności i skuteczności SARS-CoV-2 RNA SZCZEPIONKI KANDYDATÓW PRZECIW COVID-19 osób zdrowych
https://cdn.pfizer.com/pfizercom/2020-11/C4591001_Clinical_Protocol_Nov2020.pdf
3. (2021). Badanie opisujące bezpieczeństwo, tolerancję, immunogenność i skuteczność kandydatów na szczepionki RNA przeciwko COVID-19 u zdrowych osób. https://clinicaltrials.gov/ct2/show/NCT04368728
4. (2018). Krok 3: Badania kliniczne https://www.fda.gov/patients/drug-development-process/step-3-clinical-research
Czy masz świadomość, że ludzie przez setki lat leczyli się korzystając z osiągnięć medycyny naturalnej?Czy opierasz swoje rozumowanie na logice i faktach, ciągle poszukując nowych informacji?
When you subscribe to the blog, we will send you an e-mail when there are new updates on the site so you wouldn't miss them.
Komentarze